Białaczka u dzieci
Od przyczyn i diagnozy po nowoczesne metody leczenia
Od wielkiej niewiadomej do sukcesu współczesnej medycyny
Białaczka dziecięca, która jeszcze kilkadziesiąt lat temu była traktowana jak wyrok śmierci, dziś stanowi jeden z największych sukcesów medycyny. Przełom, jaki dokonał się w tej dziedzinie, jest ogromny: w latach 50. XX wieku choroba ta była niemal w 100% śmiertelna. Obecnie, dzięki systematycznym badaniom klinicznym prowadzonym w wielu ośrodkach na świecie oraz lepszemu zrozumieniu biologii nowotworów, wskaźnik wyleczalności ostrej białaczki limfoblastycznej (ALL) sięga aż 90%.
Niniejsze opracowanie to kompleksowe źródło wiedzy, które łączy najnowsze odkrycia z dziedziny genetyki, odporności (immunologii) oraz nowoczesnego leczenia farmakologicznego. Szczególną uwagę poświęcono w nim polskim programom leczenia, takim jak:
PPLLSG (Polish Pediatric Leukemia and Lymphoma Study Group): To Polskie Towarzystwo Onkologii i Hematologii Dziecięcej. To nasi eksperci, którzy adaptują światowe standardy do polskich warunków i tworzą programy takie jak cALL-POL.
Warto wiedzieć, że Polska jest pełnoprawnym członkiem I-BFM-SG, co oznacza, że polskie dzieci są leczone według dokładnie tych samych, najwyższych standardów, co dzieci w Niemczech czy USA.
Czym dokładnie jest białaczka?
Aby zrozumieć tę chorobę, musimy przestać postrzegać ją jako jedno schorzenie. W rzeczywistości jest to zróżnicowana grupa nowotworów układu krwiotwórczego. Jej istotą jest niekontrolowane namnażanie się niedojrzałych, nieprawidłowych komórek w szpiku kostnym.
Ten proces niesie ze sobą poważne konsekwencje:
- Wypieranie zdrowych komórek: Nowotworowe „klony” zajmują miejsce prawidłowych krwinek, co prowadzi do niewydolności szpiku.
- Rozprzestrzenianie się choroby: Komórki nowotworowe mogą przedostawać się ze szpiku do innych narządów w całym organizmie.
Nowa era leczenia: Precyzja zamiast siły
Współczesna onkologia dziecięca przechodzi ewolucję. Choć klasyczna chemioterapia, która niszczy szybko dzielące się komórki, wciąż jest stosowana, lekarze coraz częściej korzystają z terapii celowanych molekularnie oraz immunoterapii.
Metody te pozwalają na:
- Precyzyjne uderzenie bezpośrednio w mechanizmy, które napędzają rozwój nowotworu.
- Oszczędzenie zdrowych tkanek, co ma na celu zminimalizowanie skutków ubocznych, które mogłyby ujawnić się u dziecka w przyszłości, wiele lat po zakończeniu leczenia.
Fizjologia układu krwiotwórczego i patomechanizm transformacji białaczkowej
y w pełni zrozumieć naturę białaczki, konieczne jest zgłębienie fizjologii procesu krwiotworzenia (hematopoezy), który u dziecka zachodzi głównie w jamach szpikowych kości płaskich (mostek, talerze biodrowe) oraz w nasadach kości długich.
Budowa układu krwiotwórczego: Skąd biorą się komórki krwi?
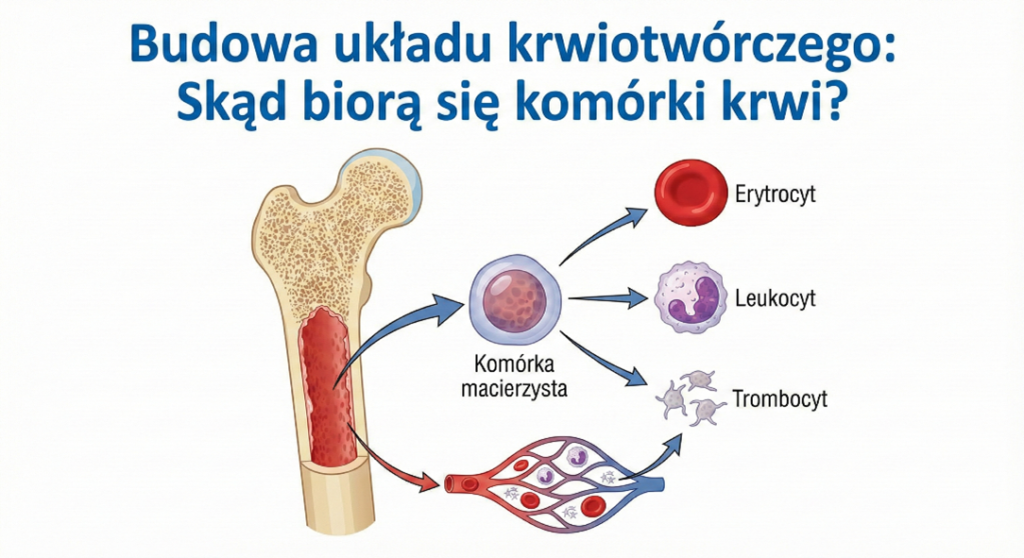
W warunkach fizjologicznych, wszystkie komórki krwi wywodzą się z multipotencjalnej krwiotwórczej komórki macierzystej (HSC – Hematopoietic Stem Cell). Komórka ta posiada unikalną zdolność do samoodnowy oraz różnicowania się w dwie główne linie:
- Linia mieloidalna: Daje początek erytrocytom (krwinkom czerwonym), trombocytom (płytkom krwi), granulocytom (neutrofile, eozynofile, bazofile) oraz monocytom.
- Linia limfoidalna: Daje początek limfocytom B (odpowiedzialnym za odporność humoralną i produkcję przeciwciał), limfocytom T (odpowiedzialnym za odporność komórkową) oraz komórkom NK (Natural Killer).
Proces ten jest ściśle regulowany przez cytokiny i czynniki wzrostu, które sterują ekspresją odpowiednich czynników transkrypcyjnych, prowadząc komórkę przez kolejne etapy dojrzewania – od blasta, przez formy pośrednie, aż do dojrzałej, funkcjonalnej komórki, która opuszcza szpik i trafia do krążenia obwodowego.
Blokada różnicowania i niekontrolowana proliferacja
Istotą białaczki jest zaburzenie tego precyzyjnego mechanizmu. W wyniku nabytych mutacji genetycznych dochodzi do zatrzymania (bloku) różnicowania komórki na wczesnym etapie rozwoju (zazwyczaj na etapie blasta). Zmutowana komórka – blast białaczkowy – nie traci jednak zdolności do podziału. Wręcz przeciwnie, zyskuje przewagę proliferacyjną nad prawidłowymi komórkami i staje się nieśmiertelna (unika apoptozy, czyli programowanej śmierci).
W ostrej białaczce limfoblastycznej (ALL) transformacji ulegają prekursory limfocytów B lub T. W ostrej białaczce szpikowej (AML) proces ten dotyczy prekursorów linii mieloidalnej.Nagromadzenie miliardów takich niefunkcjonalnych komórek w ograniczonej przestrzeni jamy szpikowej prowadzi do fizycznego wypierania prawidłowego utkania krwiotwórczego. Skutkiem tego jest pancytopenia obwodowa – brak czerwonych krwinek, płytek i dojrzałych leukocytów, co manifestuje się klinicznym obrazem choroby.
Model „Dwóch Uderzeń” (Two-Hit Hypothesis)
Współczesna genetyka molekularna sugeruje, że rozwój białaczki u dzieci jest często procesem wieloetapowym.
- Pierwsze uderzenie (First Hit): Często następuje jeszcze w życiu płodowym (in utero). Powstają wówczas tzw. fuzje genowe inicjujące, takie jak ETV6-RUNX1 (t(12;21)). Badania krwi pępowinowej wykazują, że nawet 1% noworodków posiada komórki z tą translokacją, jednak zachorowalność na białaczkę jest znacznie niższa (ok. 1 na 2000 dzieci z tą zmianą). Świadczy to o tym, że pierwsza mutacja tworzy jedynie stan przedbiałaczkowy (pre-leukemic clone).
- Drugie uderzenie (Second Hit): Aby rozwinęła się pełnoobjawowa choroba, konieczne są dodatkowe zdarzenia mutagenne, które zachodzą już po urodzeniu. Mogą one obejmować delecje genów supresorowych (np. IKZF1, CDKN2A) lub mutacje aktywujące szlaki sygnałowe (np. kinazy FLT3, szlak RAS).
Epidemiologia i etiologia: Poszukiwanie przyczyn
W Polsce rocznie rozpoznaje się około 1100-1200 nowych nowotworów u dzieci, z czego białaczki stanowią najliczniejszą grupę (ok. 26-30%).Zapadalność szacuje się na około 4 przypadki na 100 000 dzieci rocznie. Szczyt zachorowań na ALL przypada na wiek przedszkolny (2-5 lat), co wiąże się z dynamicznym rozwojem układu immunologicznego w tym okresie. W przypadku AML rozkład wiekowy jest bardziej równomierny, z lekką przewagą u niemowląt i nastolatków.
Czynniki genetyczne i zespoły predysponujące
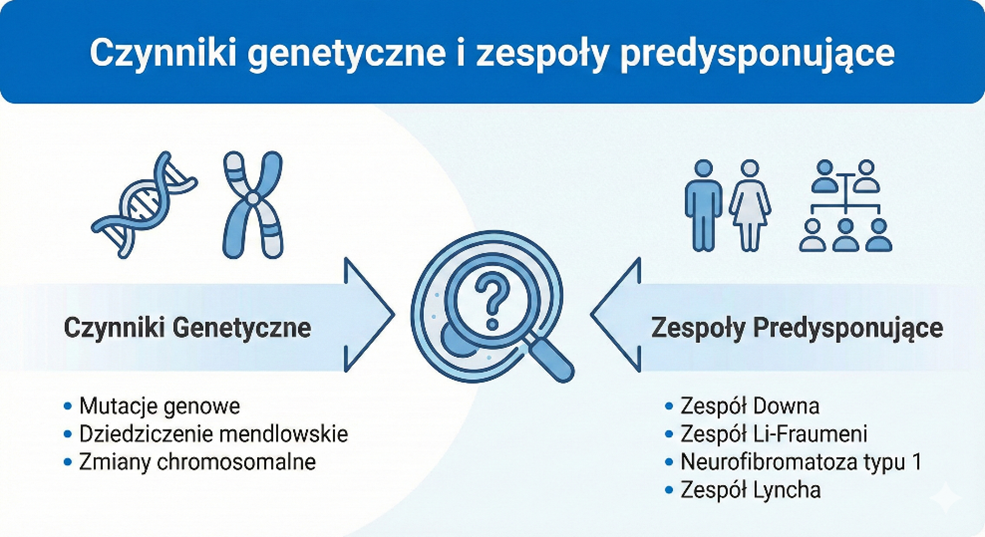
Choć większość przypadków białaczki powstaje de novo (sporadycznie), u około 5-10% pacjentów można zidentyfikować wrodzone zespoły predysponujące do nowotworzenia:
- Zespół Downa (Trisomia 21): Ryzyko białaczki jest tu 50-150 razy wyższe. Dzieci te mają specyficzny profil biologiczny choroby (często AML-M7) i wymagają zmodyfikowanych protokołów leczenia ze względu na wyższą toksyczność leków.
- Zespół Li-Fraumeni: Mutacja w genie TP53, „strażniku genomu”, prowadzi do dramatycznego wzrostu ryzyka wielu nowotworów, w tym białaczek.
- Zespoły nieprawidłowej naprawy DNA: Ataksja-teleangiektazja, Zespół Blooma, Niedokrwistość Fanconiego.
Hipoteza „Opóźnionej Infekcji” (Greaves’ Hypothesis)
W kontekście czynników środowiskowych, najsilniejszą pozycję naukową posiada teoria prof. Mela Greavesa. Sugeruje ona, że ostra białaczka limfoblastyczna (szczególnie podtyp cALL) jest paradoksalnym skutkiem postępu cywilizacyjnego i nadmiernej higieny.
Układ odpornościowy niemowlęcia pozbawiony kontaktu z drobnoustrojami w pierwszym okresie życia (izolacja, brak rodzeństwa, brak kontaktu z rówieśnikami) pozostaje „niewytrenowany”. Gdy takie dziecko trafi do przedszkola i zetknie się z falą powszechnych infekcji, jego układ immunologiczny reaguje w sposób dysregulowany, co u dzieci z obecnym klonem pre-białaczkowym (pierwsze uderzenie) prowokuje powstanie wtórnych mutacji (drugie uderzenie) i rozwój białaczki. Badania epidemiologiczne potwierdzają, że wczesne posłanie dziecka do żłobka może pełnić funkcję ochronną.
Inne czynniki środowiskowe
Rodzice często pytają o wpływ diety, smogu czy pól elektromagnetycznych.
- Promieniowanie jonizujące: Jest udowodnionym czynnikiem ryzyka (np. po wybuchach jądrowych, w radioterapii), ale dawki diagnostyczne RTG (np. zdjęcie zęba) nie mają istotnego wpływu statystycznego.
- Pola elektromagnetyczne: Wielkie badania populacyjne nie potwierdziły jednoznacznie związku między mieszkaniem w pobliżu linii wysokiego napięcia a białaczką, choć temat ten wciąż budzi kontrowersje badawcze.
- Czynniki chemiczne: Ekspozycja na benzen, pestycydy czy niektóre chemioterapeutyki (leki alkilujące, inhibitory topoizomerazy II) może prowadzić do wtórnych białaczek (t-AML/t-ALL).
Klasyfikacja i charakterystyka podtypów białaczek
Precyzyjna klasyfikacja jest fundamentem doboru terapii. Historyczny podział morfologiczny FAB (Francusko-Amerykańsko-Brytyjski) L1-L3 został zastąpiony przez nowoczesną klasyfikację WHO, opartą na immunofenotypie i genetyce.
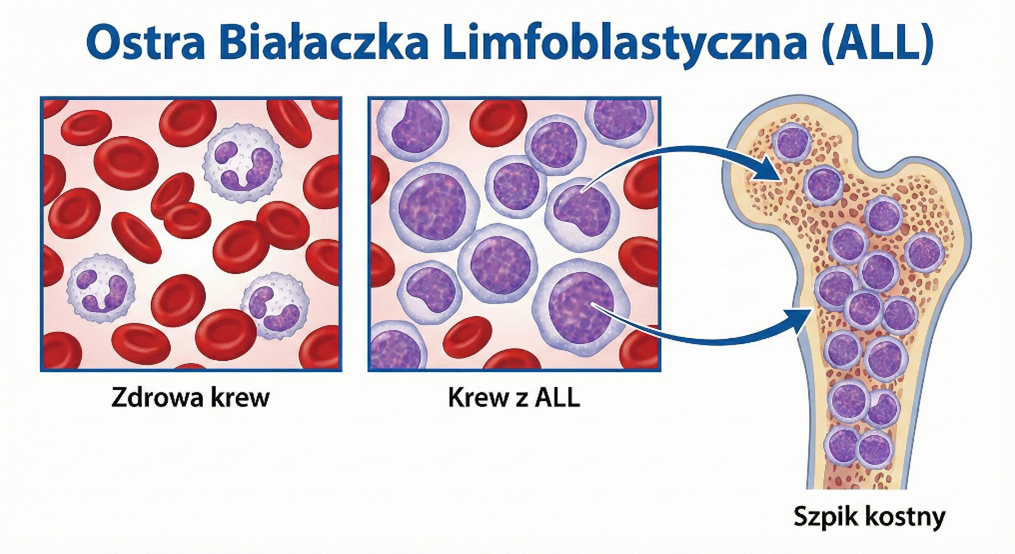
Ostra Białaczka Limfoblastyczna (ALL)
Stanowi ok. 80% wszystkich białaczek dziecięcych.
- B-komórkowa ALL (B-ALL): Najczęstsza postać (ok. 85% ALL). Wywodzi się z prekursorów linii B. Dzieli się na podtypy: pro-B, common-B (najczęstszy), pre-B. Rokowanie jest zazwyczaj dobre, choć zależy silnie od profilu genetycznego.
- T-komórkowa ALL (T-ALL): Stanowi ok. 15% ALL. Częściej dotyczy starszych chłopców. Charakteryzuje się wysoką leukocytozą i często obecnością guza w śródpiersiu (powiększona grasica), co może prowadzić do zespołu żyły głównej górnej. Wymaga bardziej intensywnego leczenia.
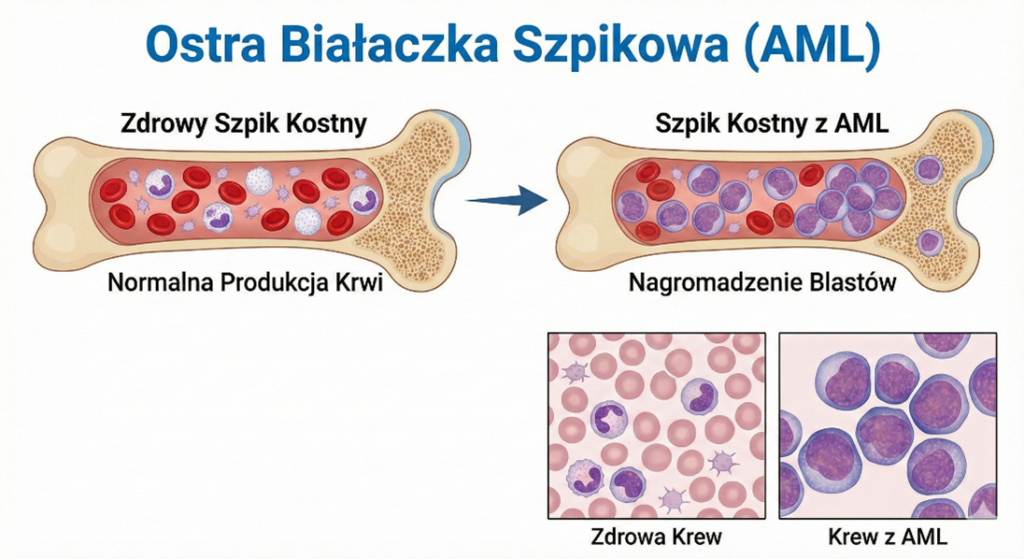
Ostra Białaczka Szpikowa (AML)
Stanowi ok. 15-20% przypadków. Jest chorobą znacznie trudniejszą w leczeniu. Komórki białaczkowe wywodzą się z linii mieloidalnej (granulocytarnej, monocytarnej, erytroidalnej lub megakariocytarnej). Klasyfikacja WHO wyróżnia wiele podtypów opartych na zmianach genetycznych, np. AML z t(8;21), AML z inv(16), czy ostra białaczka promielocytowa (APL) z t(15;17) – ta ostatnia leczona jest odmiennie, z użyciem kwasu all-trans retinowego (ATRA).
Białaczki Niemowlęce (Infant Leukemia)
Dotyczą dzieci poniżej 1. roku życia. Są biologicznie odrębne, często związane z rearanżacją genu KMT2A (dawniej MLL) na chromosomie 11q23. Cechują się bardzo agresywnym przebiegiem, opornością na standardowe leki i wysokim ryzykiem nawrotu, często wymagają przeszczepienia szpiku w pierwszej remisji.
Symptomatologia kliniczna: „Wielki Maskarator”
Objawy białaczki wynikają bezpośrednio z patomechanizmu choroby: wyparcia prawidłowego szpiku i naciekania narządów. Początek może być nagły lub podstępny, trwający kilka tygodni.
Triada objawów niewydolności szpiku
- Niedokrwistość (Anemia): Wynika ze spadku produkcji erytrocytów. Dziecko staje się nienaturalnie blade (woskowe), apatyczne, traci apetyt. Starsze dzieci zgłaszają bóle głowy, szum w uszach, nietolerancję wysiłku. Rodzice często zauważają, że dziecko „przestało biegać” na placu zabaw.
- Małopłytkowość (Trombocytopenia): Niedobór płytek krwi prowadzi do skazy krwotocznej. Charakterystyczne są wybroczyny (petocje) – drobne, czerwone punkciki na skórze, nieblednące pod uciskiem, przypominające wysypkę. Pojawiają się siniaki w nietypowych miejscach (brzuch, plecy, uszy) bez urazu, krwawienia z nosa, dziąseł.
- Neutropenia: Mimo często podwyższonej liczby białych krwinek w morfologii (leukocytoza), są to komórki nowotworowe, niezdolne do walki z infekcjami. Funkcjonalnych neutrofili jest skrajnie mało. Prowadzi to do nawracających, ciężkich infekcji, gorączek niewiadomego pochodzenia, zapaleń jamy ustnej, angin martwiczych, które nie reagują na standardowe antybiotyki.
Objawy proliferacyjne (naciekowe)
- Bóle kostno-stawowe: Spowodowane rozprężaniem jamy szpikowej przez gwałtownie dzielące się blasty. Często mylone z bólami wzrostowymi lub reumatycznymi. Ból może być tak silny, że dziecko przestaje chodzić, kuleje lub budzi się z krzykiem w nocy.
- Powiększenie narządów (Organomegalia): Nacieki blastów w wątrobie i śledzionie powodują powiększenie obwodu brzucha, uczucie pełności, bóle brzucha.
- Limfadenopatia: Powiększenie węzłów chłonnych (szyjnych, pachowych, pachwinowych), które są zazwyczaj niebolesne, twarde i zbijają się w pakiety.
- Objawy ze strony Ośrodkowego Układu Nerwowego (Neuroleukemia): Gdy białaczka zajmie opony mózgowo-rdzeniowe, pojawiają się poranne bóle głowy, wymioty (chlustające), zaburzenia widzenia, porażenia nerwów czaszkowych (np. opadanie kącika ust).
- Zespół żyły głównej górnej: W T-ALL powiększone węzły śródpiersia mogą uciskać żyłę główną, powodując obrzęk twarzy i szyi, duszność, kaszel. Jest to stan zagrożenia życia.
Objawy specyficzne dla AML
- Przerost dziąseł: Nacieki komórek monocytarnych w dziąsłach, które stają się obrzęknięte, bolesne i krwawiące.
- Chloroma (Mięsak granulocytarny): Guz lity zbudowany z komórek białaczkowych, mogący występować np. w oczodole (wytrzeszcz gałki ocznej) lub skórze.
- Leukemia cutis: Nacieki skórne w postaci twardych guzków.
Diagnostyka laboratoryjna i obrazowa: Ścieżka do rozpoznania
Proces diagnostyczny musi być szybki i precyzyjny. Jego celem jest nie tylko potwierdzenie nowotworu, ale przede wszystkim uzyskanie pełnego profilu biologicznego choroby, co determinuje stratyfikację ryzyka.
Morfologia krwi obwodowej
Jest pierwszym sygnałem alarmowym. Typowy obraz to:
- Leukocytoza: Często bardzo wysoka (ponad 20-50 tys./µl), choć u części pacjentów liczba leukocytów może być w normie lub obniżona (leukopenia).
- Niedokrwistość i małopłytkowość.
- Obecność blastów: W rozmazie ręcznym widoczne są niedojrzałe komórki, które normalnie nie występują we krwi obwodowej. Uwaga: Automatyczne analizatory mogą błędnie zaklasyfikować blasty jako monocyty lub limfocyty, dlatego kluczowa jest ocena mikroskopowa przez doświadczonego diagnostę.
Biopsja i trepanobiopsja szpiku kostnego
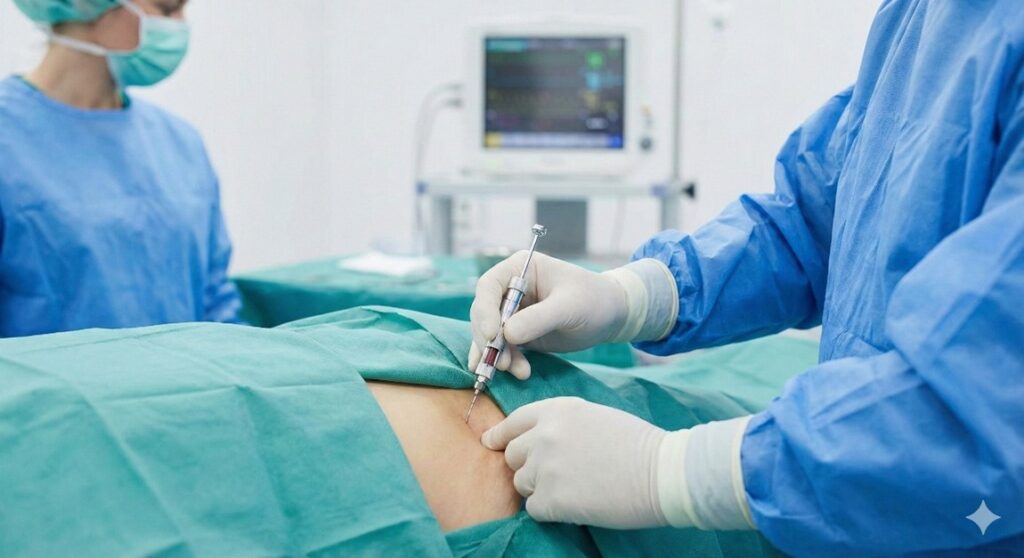
To „złoty standard” diagnostyczny. Zabieg wykonuje się w znieczuleniu ogólnym (sedacji), aby zminimalizować traumę dziecka.
- Aspiracja: Pobranie płynnego szpiku (zazwyczaj z talerza kości biodrowej) do badań cytologicznych, immunofenotypowych i genetycznych.
- Trepanobiopsja: Pobranie fragmentu kości ze szpikiem. Konieczna, gdy szpik jest tak gęsto upakowany komórkami („packed marrow”), że nie udaje się go zaaspirować (tzw. „puste ukłucie” – dry tap).
Rozpoznanie białaczki stawia się zazwyczaj, gdy odsetek blastów w szpiku przekracza 20% (według WHO), choć w praktyce często wynosi on 80-90%.
Nakłucie lędźwiowe (Lumbopunkcja)
Pobranie płynu mózgowo-rdzeniowego w celu oceny zajęcia OUN (status CNS). W płynie poszukuje się komórek białaczkowych (cytologia). Wynik dodatni (CNS-2 lub CNS-3) zmienia klasyfikację ryzyka i wymaga intensywniejszego leczenia dokanałowego.
Immunofenotypowanie (Cytometria przepływowa)
Metoda ta pozwala na szybkie (w kilka godzin) określenie, z jakiej linii wywodzą się komórki białaczkowe. Wykorzystuje się przeciwciała znakowane fluorochromami, które wiążą się ze specyficznymi antygenami na powierzchni komórek (CD – Cluster of Differentiation).
- Markery B-ALL: CD19, CD22, CD79a, CD10 (CALLA), PAX5.
- Markery T-ALL: CD3 (cytoplazmatyczne i powierzchniowe), CD7, CD5.
- Markery Mieloidalne (AML): CD13, CD33, MPO (mieloperoksydaza – kluczowa dla AML).
Badania obrazowe i biochemiczne
- RTG klatki piersiowej: Ocena obecności guza śródpiersia.
- USG jamy brzusznej: Ocena wielkości wątroby, śledziony i nerek.
- Biochemia: Ocena funkcji nerek i wątroby. Kluczowy jest poziom kwasu moczowego i LDH (dehydrogenaza mleczanowa), które są markerami rozpadu guza i obrotu komórkowego.
Stratyfikacja ryzyka i genetyka molekularna: Klucz do precyzji
Współczesne protokoły leczenia, takie jak cALL-POL, opierają się na dostosowaniu intensywności terapii do ryzyka wznowy. O przydziale do grupy ryzyka (Standardowe – SR, Pośrednie – IR, Wysokie – HR) decydują dwa główne filary: Genetyka i Odpowiedź na leczenie.
Badania genetyczne (cytogenetyczne i molekularne)
Badania genetyczne są dziś kluczowym elementem diagnozy białaczki u dzieci. To one pozwalają lekarzom ocenić, jak choroba zareaguje na leczenie i czy dany pacjent potrzebuje standardowej terapii, czy bardziej agresywnego podejścia.
Poniżej znajdziesz szczegółowe omówienie zmian w genach (aberracji), podzielone na grupy w zależności od tego, jakie dają szanse na wyleczenie.
1. Zmiany o bardzo dobrym rokowaniu (Największa szansa na wyleczenie)
W tej grupie znajdują się dzieci, u których standardowe leczenie zazwyczaj przynosi świetne efekty.
- Fuzja genów t(12;21) ETV6-RUNX1: Występuje u co czwartego dziecka z białaczką typu B-ALL (25%). Pacjenci ci bardzo dobrze reagują na L-asparaginazę (jeden z głównych leków w chemioterapii). Rokowania są określane jako bardzo dobre.
- Wysoka hiperdiploidia (powyżej 50 chromosomów): To stan, w którym komórka nowotworowa ma znacznie więcej chromosomów niż zdrowa. Dotyczy to około 25–30% przypadków B-ALL. Te komórki są niezwykle wrażliwe na metotreksat, co przekłada się na bardzo dobre wyniki leczenia.
2. Zmiany o dobrym lub pośrednim rokowaniu
- t(8;21) lub inv(16) (tzw. CBF-AML): Zmiany charakterystyczne dla ostrej białaczki szpikowej (AML), występujące u co czwartego pacjenta (25%). Rokowanie jest dobre, ponieważ komórki te wykazują wysoką wrażliwość na lek o nazwie cytarabina.
- t(9;22) BCR-ABL1 (tzw. chromosom Philadelphia): Występuje u 3–5% dzieci z B-ALL. Dawniej było to rokowanie bardzo złe, ale dzięki nowoczesnej medycynie zmieniło się na pośrednie. Kluczem do sukcesu jest dodanie do leczenia tzw. inhibitorów kinaz (np. imatynibu), które precyzyjnie blokują białko wywołujące chorobę.
3. Zmiany o złym rokowaniu (Wysokie ryzyko)
W tych przypadkach lekarze muszą stosować intensywniejsze metody, często od razu planując przeszczep szpiku.
- Białaczka „Ph-like” (Philadelphia-like): Występuje u ok. 15% pacjentów z B-ALL. Genetycznie przypomina białaczkę z chromosomem Philadelphia, ale mechanizm jest nieco inny. Często towarzyszą jej zmiany w genie IKZF1. Wymaga leczenia celowanego nowoczesnymi lekami (np. ruksolitynibem).
- Rearanżacje KMT2A (MLL): Dotyczą aż 80% niemowląt chorych na białaczkę. To bardzo agresywna postać choroby, która często nie reaguje na sterydy, a rokowania są niestety złe.
- Hipodiploidia (poniżej 44 chromosomów): Rzadka zmiana w B-ALL, ale o bardzo złym rokowaniu. Często wiąże się z mutacjami w genie TP53 i jest bezpośrednim wskazaniem do przeszczepu szpiku kostnego (HSCT).
- iAMP21 (amplifikacja chromosomu 21): Występuje u ok. 2% pacjentów z B-ALL. Rokowanie jest złe, dlatego dzieci z tą zmianą są od początku leczone w grupach wysokiego ryzyka (HR) przy użyciu intensywnej terapii.
- Monosomia 7 lub del(5q): Rzadkie zmiany w białaczkach szpikowych (AML). Rokowanie jest złe, a choroba często wywodzi się z wcześniejszych zaburzeń szpiku (MDS). Tutaj również konieczny jest zazwyczaj przeszczep szpiku.
Minimalna Choroba Resztkowa (MRD – Minimal Residual Disease)
Jest to najważniejszy niezależny czynnik rokowniczy. Określa ilość komórek białaczkowych, które przetrwały w organizmie po wstępnym leczeniu.
- Badanie wykonuje się w ściśle określonych punktach czasowych: np. w 15. i 33. dniu leczenia (dla ALL).
- Metody: Cytometria przepływowa (czułość 10^-4) lub PCR (czułość 10^-5).
- Interpretacja:
MRD ujemne (<0.01%): Doskonała odpowiedź, pacjent może być leczony mniej toksycznym protokołem (deeskalacja).
MRD dodatnie: Świadczy o chemiooporności. Pacjent automatycznie trafia do grupy wysokiego ryzyka (HR) i otrzymuje intensywniejsze leczenie, aby zapobiec wznowie.2
Strategie terapeutyczne: Ostra Białaczka Limfoblastyczna (ALL)
Leczenie ALL w Polsce w ramach grupy cALL-POL (bazującej na protokole AIEOP-BFM-2017) trwa łącznie około 2 lat i jest podzielone na etapy.
Faza 1: Indukcja remisji (Protokół I)
- Cel: Redukcja masy guza o 99% (z 10^12 do poniżej 10^9 komórek) i przywrócenie prawidłowego krwiotworzenia.
- Kluczowe leki:
- Glikokortykosteroidy (Prednizon/Deksametazon): Stanowią fundament leczenia ALL. Działają proapoptycznie na limfoblasty. Wstępna odpowiedź na sterydy (liczba blastów we krwi w 8. dobie) jest ważnym czynnikiem prognostycznym („Dobra odpowiedź sterydowa” vs „Zła odpowiedź sterydowa”).
- Winkrystyna: Alkaloid barwinka, hamuje podziały komórkowe.
- L-asparaginaza (PEG-Asparaginaza): Enzym rozkładający asparaginę – aminokwas niezbędny komórkom białaczkowym do życia (prawidłowe komórki potrafią go same syntezować, białaczkowe nie).
- Antracykliny (Daunorubicyna): Dodawane u pacjentów z grup pśredniego i wysokiego ryzyka.
- Czas: Ok. 4-6 tygodni.
- Powikłania: Ryzyko zakrzepicy, zapalenia trzustki (po asparaginazie), polineuropatii (po winkrystynie).
Faza 2: Konsolidacja (Protokół IB)
- Cel: Eliminacja komórek lekoopornych, które przeżyły indukcję.
- Strategia: Wprowadzenie nowych leków, aby uniknąć oporności krzyżowej: Cyklofosfamid, Cytarabina (Ara-C), 6-Merkaptopuryna. Jest to faza intensywnej „rotacji” leków.
Faza 3: Profilaktyka i leczenie OUN (Ośrodkowego Układu Nerwowego)
Bariera krew-mózg chroni komórki białaczkowe przed większością leków dożylnych. Bez celowanego leczenia, OUN staje się „sanktuarium” dla białaczki, co prowadzi do nawrotów.
- Metoda: Dokanałowe podawanie leków (Metotreksat, Cytarabina, Sterydy) bezpośrednio do płynu mózgowo-rdzeniowego poprzez punkcje lędźwiowe.
- Radioterapia: Obecnie ograniczana tylko do wybranych pacjentów z grupy najwyższego ryzyka (z masywnym zajęciem OUN lub T-ALL wysokiego ryzyka), ze względu na odległe skutki neurotoksyczne.2
Faza 4: Reindukcja (Protokół II)
Powtórzenie intensywnego leczenia indukcyjnego po kilku miesiącach. Ma na celu ostateczne „oczyszczenie” organizmu z resztek choroby przed fazą podtrzymującą. Jest to okres bardzo trudny dla pacjenta, związany z wysokim ryzykiem powikłań infekcyjnych.
Faza 5: Leczenie podtrzymujące (Maintenance)
- Cel: Utrzymanie remisji i eliminacja „uśpionych” komórek białaczkowych.
- Forma: Leczenie ambulatoryjne (domowe). Codziennie doustnie 6-Merkaptopuryna i raz w tygodniu Metotreksat.
- Czas: Do osiągnięcia 104 tygodni od rozpoczęcia leczenia. W tym czasie dziecko powoli wraca do normalnego życia, szkoły, choć nadal wymaga monitorowania morfologii i prób wątrobowych.
Strategie terapeutyczne: Ostra Białaczka Szpikowa (AML)
Leczenie AML różni się fundamentalnie od ALL. Jest krótsze (ok. 5-6 miesięcy), ale znacznie bardziej intensywne („myeloablacyjne”). W Polsce stosuje się protokół AIEOP-AML-BFM 2020.
Specyfika leczenia AML
- Indukcja: Oparta na Cytarabinie i Antracyklinach (tzw. schemat „7+3” lub jego modyfikacje: blok ADE, liposomalna daunorubicyna). Leki te doszczętnie niszczą szpik, powodując głęboką aplazję trwającą 3-4 tygodnie. W tym czasie pacjent jest całkowicie pozbawiony odporności i wymaga izolacji oraz intensywnego leczenia wspomagającego.
- Konsolidacja: Kolejne bloki chemioterapii z wysokimi dawkami cytarabiny (HiDAC), mające na celu prewencję nawrotu.
- Brak fazy podtrzymującej: W przeciwieństwie do ALL, leczenie podtrzymujące w AML (poza podtypem APL) nie przynosi korzyści.
Wskazania do HSCT: W AML znacznie częściej wykonuje się przeszczepienie szpiku w pierwszej remisji, szczególnie u pacjentów z grupy wysokiego ryzyka genetycznego lub wolną odpowiedzią na leczenie.
Immunoterapia i terapie celowane: Nowa era w leczeniu oporności
Dla pacjentów z chorobą oporną, nawrotową lub z grup najwyższego ryzyka, klasyczna chemia często jest niewystarczająca. Z pomocą przychodzi inżynieria genetyczna i immunologia.
Przeciwciała monoklonalne i bispecyficzne
- Blinatumomab (BiTE – Bispecific T-cell Engager): Innowacyjny lek, który działa jak „adapter”. Jednym końcem wiąże się z antygenem CD19 na komórce białaczkowej, a drugim z antygenem CD3 na limfocycie T pacjenta. Prowadzi to do aktywacji limfocytu T i zniszczenia komórki nowotworowej (efekt „pocałunku śmierci”). W protokole cALL-POL jest on wprowadzany u pacjentów HR jako terapia pomostowa lub zastępująca toksyczne bloki chemii, co znacząco poprawia rokowanie.
- Inotuzumab ozogamycyny: Przeciwciało anty-CD22 sprzężone z silną toksyną (kalicheamycyną). Lek wiąże się z komórką białaczkową, wnika do jej wnętrza i uwalnia toksynę, która niszczy DNA. Stosowany w nawrotach ALL.
- Gemtuzumab ozogamycyny: Analogiczny mechanizm dla antygenu CD33 w komórkach AML. Został włączony do standardu leczenia niektórych postaci AML w protokole AIEOP-AML-BFM 2020.
Terapia CAR-T (Chimeric Antigen Receptor T-cells)
Jest to najbardziej zaawansowana forma immunoterapii, dostępna w Polsce (np. preparat Kymriah) dla dzieci z oporną/nawrotową B-ALL.
- Mechanizm: Od pacjenta pobiera się jego własne limfocyty T (w procesie leukaferezy). W laboratorium genetycznym wprowadza się do nich gen kodujący receptor CAR, który rozpoznaje antygen CD19. Tak zmodyfikowane „żywe leki” są namnażane i podawane z powrotem pacjentowi.
- Efekt: Komórki CAR-T potrafią krążyć w organizmie latami, aktywnie wyszukując i niszcząc komórki białaczkowe. Skuteczność tej metody w grupach beznadziejnych sięga 80% remisji całkowitych.
Inhibitory Kinaz Tyrozynowych (TKI)
Dla pacjentów z białaczką Ph+ (BCR-ABL1) lub Ph-like, dodanie do chemioterapii leków takich jak Imatynib lub Dasatynib jest standardem. Blokują one specyficzne białka sygnałowe napędzające wzrost nowotworu. Nowsze badania (Rux-ALL) testują Ruxolitinib (inhibitor JAK1/2) dla pacjentów ze zmianami w szlaku JAK-STAT.
Przeszczepienie Komórek Krwiotwórczych (HSCT)
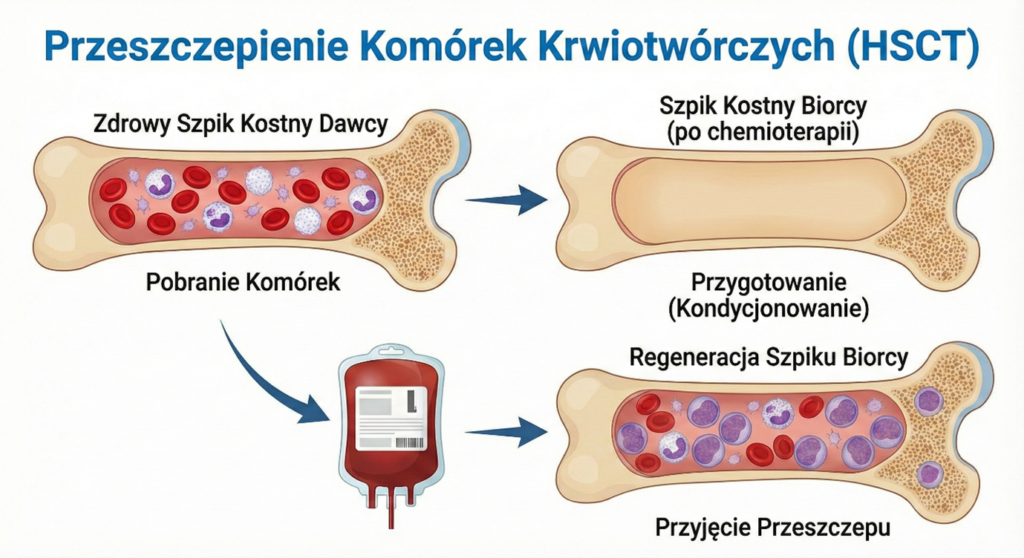
Przeszczepienie (potocznie „szpiku”) jest procedurą ratującą życie, ale obarczoną wysokim ryzykiem śmiertelności okołoprzeszczepowej (TRM), dlatego kwalifikacja jest bardzo ostrożna.
Rodzaje przeszczepień:
- Allogeniczne (allo-HSCT): Od dawcy (rodzinnego lub niespokrewnionego). Jest preferowaną metodą w białaczkach, ponieważ poza efektem wysokich dawek chemii (kondycjonowania), wykorzystuje zjawisko GvL (Graft-versus-Leukemia) – nowy układ odpornościowy dawcy rozpoznaje i niszczy resztki białaczki.
- Autologiczne: Obecnie rzadko stosowane w białaczkach dziecięcych (głównie w niektórych guzach litych), gdyż nie zapewnia efektu GvL.
Wskazania:
Większość nawrotów (wznowy) choroby.
Brak remisji po leczeniu indukcyjnym.
Wysoki poziom choroby resztkowej (MRD) po konsolidacji.
Białaczki o bardzo niekorzystnym profilu genetycznym (np. hipodiploidia, niemowlęca ALL z rearanżacją KMT2A).
Leczenie wspomagające i opieka pielęgniarska: Domowy front walki
Rola rodziców w procesie leczenia jest nie do przecenienia. To oni stają się pierwszą linią obrony przed powikłaniami.
Dostęp naczyniowy centralny
Dzieciom rutynowo zakłada się cewniki centralne, aby oszczędzić żyły obwodowe i zapewnić pewną drogę podania leków drażniących.
- Cewnik tunelizowany (Broviac/Hickman): Wężyk wyprowadzony na zewnątrz klatki piersiowej. Wymaga sterylnej pielęgnacji, płukania heparyną i ochrony przed zamoczeniem. Jest preferowany przy intensywnej terapii (częste przetoczenia, pobrania krwi).
- Port naczyniowy (V-Port): Komora wszyta pod skórę. Wymaga nakłucia igłą Hubera. Zapewnia większy komfort (możliwość kąpieli po wyjęciu igły), ale każdorazowe podłączenie wiąże się z ukłuciem.24
Dieta neutropeniczna („Ubogobakterijna”)
Gdy poziom neutrofili spada (często do zera), przewód pokarmowy staje się wrotami zakażenia.
- Zasada: Eliminacja surowych produktów, które mogą zawierać bakterie (Listeria, Salmonella, E. coli) lub grzyby.
- Produkty zakazane: Surowe mięso, ryby, jaja na miękko, sery pleśniowe, jogurty naturalne z żywymi kulturami bakterii (chyba że pasteryzowane), orzechy (ryzyko pleśni), surowe owoce i warzywa (chyba że można je sparzyć i obrać z grubej skóry – np. banan).
- Obróbka: Gotowanie, pieczenie, duszenie. Woda tylko przegotowana lub butelkowana ze sprawdzonego źródła (świeżo otwarta).25
Higiena jamy ustnej
Chemioterapia niszczy śluzówki, powodując bolesne zapalenie (mucositis). Konieczne jest regularne płukanie jamy ustnej (preparatami odkażającymi, roztworem sody, Caphosolem), używanie miękkich szczoteczek i pędzlowanie zmian lekami przeciwgrzybiczymi (Nystatyna). Zaniedbanie higieny może prowadzić do sepsy odogniskowej.
Powikłania wczesne i toksyczność leczenia
Intensywne leczenie niesie za sobą szereg działań niepożądanych, które wymagają natychmiastowej reakcji.
Zespół Rozpadu Guza (TLS – Tumor Lysis Syndrome)
Na początku leczenia (pre-faza sterydowa), masowy rozpad komórek białaczkowych uwalnia do krwi potas, fosforany i kwas moczowy. Może to doprowadzić do ostrej niewydolności nerek i zatrzymania krążenia.
- Zapobieganie: Hiperhydratacja (nawadnianie) i leki obniżające poziom kwasu moczowego (Allopurinol, Rasburykaza).
Infekcje w neutropenii
Gorączka u dziecka z neutropenią (>38°C) jest stanem bezpośredniego zagrożenia życia. Wymaga natychmiastowego podania antybiotyków szerokowidmowych w warunkach szpitalnych, gdyż sepsa może rozwinąć się w ciągu kilku godzin.
Specyficzne toksyczności narządowe
- Winkrystyna: Neurotoksyczność obwodowa (zaparcia, niedrożność jelit, opadanie stóp, bóle szczęki).
- Asparaginaza: Zaburzenia krzepnięcia (zakrzepica lub krwawienia), ostre zapalenie trzustki, reakcje alergiczne.
- Antracykliny: Kardiotoksyczność (uszkodzenie mięśnia sercowego).
- Sterydy: Cukrzyca posteroidowa, nadciśnienie, miopatia (osłabienie mięśni), zmiany nastroju, martwica kości.
Odległe następstwa i jakość życia (Survivorship)
Wyleczenie z białaczki to dopiero połowa sukcesu. Celem współczesnej onkologii jest „Cure at the lowest cost” – wyleczenie przy minimalnych kosztach zdrowotnych. Ozdrowieńcy wymagają dożywotniego monitorowania w poradniach odległych następstw.
Aspekty psychosocjalne
Choroba dziecka jest traumą dla całej rodziny (PTSD u rodziców). Powrót do „normalności” bywa trudny. Dzieci mogą mieć problemy z rówieśnikami (wygląd, absencja w szkole) i lękiem przed nawrotem. Wsparcie psychoonkologa jest integralną częścią leczenia.
Podsumowanie i perspektywy
Białaczka u dzieci jest chorobą niezwykle złożoną, ale też chorobą, w której medycyna odniosła spektakularny sukces. Dzięki programom takim jak cALL-POL i współpracy międzynarodowej, polskie dzieci mają dostęp do terapii na światowym poziomie, w tym do najnowocześniejszych leków immunologicznych i celowanych molekularnie.
Przyszłość leczenia zmierza w kierunku „chemioterapii wolnej od chemii” (chemo-free regimens), gdzie toksyczne cytostatyki zastępowane są precyzyjnymi „inteligentnymi lekami”. Dla rodziców kluczowa jest świadomość, że choć droga leczenia jest długa i wyboista, jasny cel – całkowite wyleczenie dziecka i jego powrót do pełni życia – jest statystycznie wysoce prawdopodobny. Wiedza o chorobie, zrozumienie mechanizmów leczenia i ścisła współpraca z zespołem medycznym są najlepszą bronią, jaką rodzice mogą wyposażyć swoje dziecko w tej walce.