Zrozumieć neuroblastomę: Przewodnik dla Mamy i Taty
Patogeneza i diagnostyka
Neuroblastoma, znana w polskiej terminologii medycznej jako nerwiak zarodkowy współczulny, stanowi jedno z najbardziej złożonych i niejednorodnych wyzwań w nowoczesnej onkologii dziecięcej.
Dla rodzica, który styka się z tą diagnozą, świat medyczny nagle otwiera się z całym spektrum trudnych pojęć, statystyk i procedur. Niniejsze opracowanie ma na celu nie tylko zdefiniowanie choroby, ale przede wszystkim przeprowadzenie czytelnika przez zawiłości biologii molekularnej, standardów diagnostycznych oraz wieloetapowego leczenia, które jest obecnie dostępne w Polsce i w wiodących ośrodkach zagranicznych.
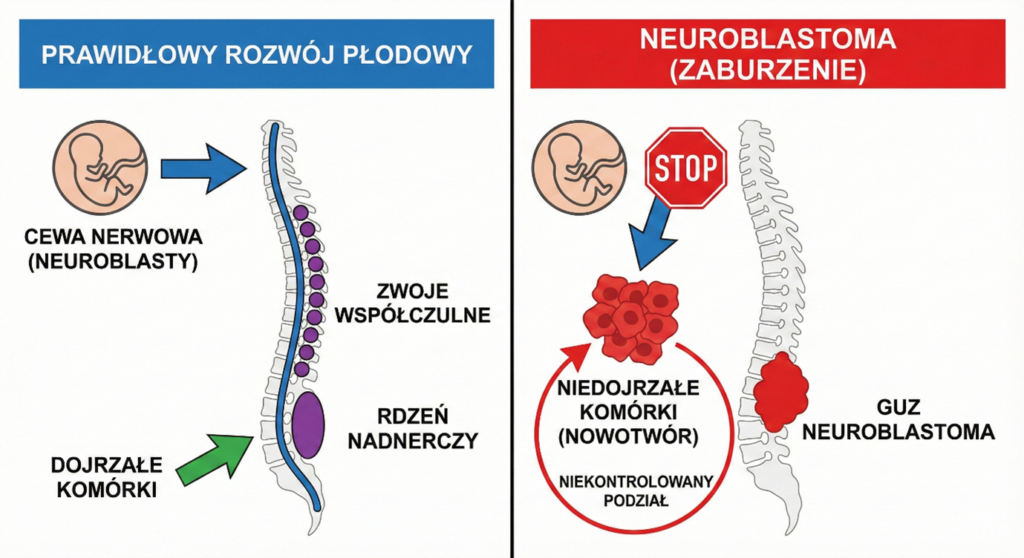
Skąd się bierze Neuroblastoma?
Jest to nowotwór wywodzący się z układu współczulnego, a dokładniej z pierwotnych komórek cewy nerwowej, zwanych neuroblastami. W prawidłowym rozwoju płodowym komórki te wędrują i dojrzewają, tworząc zwoje współczulne wzdłuż kręgosłupa oraz rdzeń nadnerczy. W przypadku neuroblastomy proces ten ulega zaburzeniu – komórki zatrzymują się na etapie niedojrzałym i zamiast pełnić swoje funkcje fizjologiczne, zaczynają się w sposób niekontrolowany dzielić.
Epidemiologia tego nowotworu wskazuje na jego wybitnie dziecięcy charakter. Jest to najczęstszy nowotwór lity występujący poza ośrodkowym układem nerwowym u dzieci, stanowiący od 7% do 10% wszystkich nowotworów wieku dziecięcego.
W Polsce każdego roku diagnozę tę słyszy rodziców około 70–80 dzieci. Co niezwykle istotne, jest to choroba „małych dzieci” – aż 90% przypadków rozpoznaje się przed ukończeniem 5. roku życia, a szczyt zachorowań przypada na okres niemowlęcy.
U dzieci powyżej 10. roku życia choroba ta jest rzadkością (2% przypadków), a u młodzieży powyżej 15 lat występuje incydentalnie (0,5%). Ta specyfika wiekowa determinuje nie tylko przebieg kliniczny, ale także biologię guza, która u niemowląt może być diametralnie inna niż u dzieci starszych.
Unikalną cechą neuroblastomy, która fascynuje naukowców i jednocześnie utrudnia rokowanie, jest jej skrajna heterogenność kliniczna. Spektrum choroby rozciąga się od guzów, które potrafią samoistnie zaniknąć (regresja) bez jakiegokolwiek leczenia – co jest zjawiskiem niemal niespotykanym w innych nowotworach złośliwych – po postacie wysoce agresywne, oporne na konwencjonalną chemioterapię i szybko dające przerzuty do kości i szpiku. Zrozumienie, dlaczego u jednego dziecka guz znika, a u drugiego wymaga najcięższego leczenia znanego medycynie, leży w genetyce komórek nowotworowych.
Biologia molekularna i uwarunkowania genetyczne
Współczesna onkologia odeszła od traktowania neuroblastomy jako jednej choroby. Obecnie wiemy, że rokowanie i wybór terapii zależą w głównej mierze od profilu genetycznego guza. To „dowód osobisty” nowotworu decyduje o tym, czy dziecko trafi do grupy obserwacji, czy zostanie zakwalifikowane do protokołu wysokiego ryzyka.
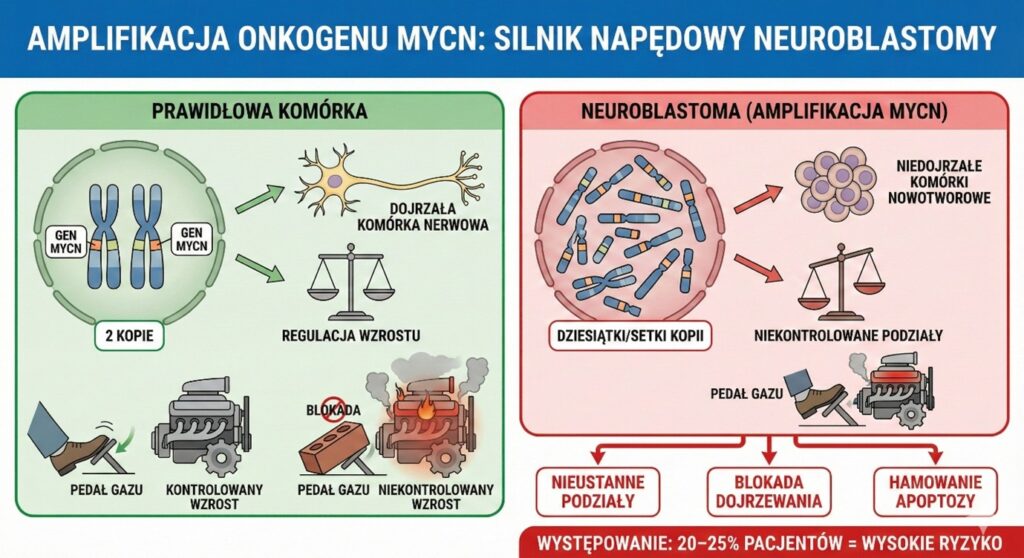
Amplifikacja onkogenu MYCN – silnik napędowy nowotworu
Najważniejszym i najlepiej zbadanym markerem w neuroblastomie jest gen MYCN. W prawidłowej komórce gen ten występuje w dwóch kopiach (po jednej od każdego z rodziców) i odpowiada za regulację wzrostu tkanki nerwowej.
W komórkach agresywnej neuroblastomy dochodzi do błędu replikacji, w wyniku którego gen ten ulega powieleniu – amplifikacji. Liczba kopii może sięgać kilkudziesięciu, a nawet kilkuset. Nadmiar białka produkowanego przez MYCN działa jak zablokowany pedał gazu w samochodzie – zmusza komórki do nieustannych podziałów, blokuje ich dojrzewanie i hamuje procesy naturalnej śmierci komórkowej (apoptozy). Amplifikacja MYCN występuje u około 20–25% pacjentów i jest niemal zawsze sygnałem alarmowym, kwalifikującym pacjenta do grupy wysokiego ryzyka, niezależnie od innych czynników. Zrozumienie tego mechanizmu było kluczowe dla opracowania strategii terapeutycznych, choć bezpośrednie farmakologiczne zablokowanie białka MYCN pozostaje wyzwaniem dla naukowców od dekad.
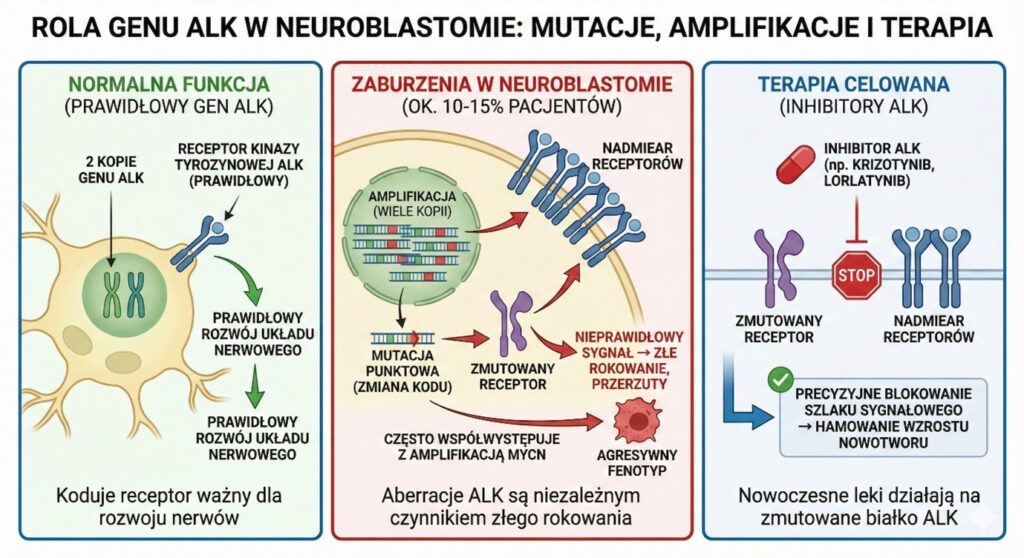
Rola genu ALK i mutacje somatyczne
Kolejnym kluczowym graczem w patogenezie neuroblastomy jest gen ALK (Anaplastic Lymphoma Kinase). Koduje on receptor kinazy tyrozynowej, który odgrywa rolę w rozwoju układu nerwowego.
Zaburzenia tego genu mogą przybierać dwie formy: mutacji punktowych (zmiana „liter” w kodzie DNA) lub amplifikacji (zwiększenie liczby kopii). Zmiany w genie ALK stwierdza się u około 10–15% pacjentów z neuroblastomą, a w rzadkich przypadkach występowania rodzinnego neuroblastomy, to właśnie mutacje ALK są dziedziczone.
Badania przeprowadzone m.in. w ramach grupy SIOPEN wykazały, że obecność aberracji w genie ALK jest niezależnym czynnikiem złego rokowania. Pacjenci z amplifikacją ALK mają statystycznie gorsze wskaźniki przeżycia całkowitego (OS), co jest szczególnie widoczne w grupie z chorobą przerzutową. Co ciekawe, amplifikacja ALK niemal zawsze współwystępuje z amplifikacją MYCN, tworząc wyjątkowo agresywny fenotyp nowotworu. Jednak odkrycie to ma również pozytywny aspekt – obecność zmutowanego białka ALK daje cel dla nowoczesnych leków, tzw. inhibitorów kinazy ALK (np. krizotynib, lorlatynib), które mogą precyzyjnie blokować ten szlak sygnałowy.
Aberracje chromosomalne i inne markery
Oprócz mutacji pojedynczych genów, komórki neuroblastomy często wykazują duże zmiany w strukturze chromosomów. Do tzw. segmentowych aberracji chromosomalnych (SCA), które wiążą się z gorszym rokowaniem, należą:
- Delecja 1p: Utrata fragmentu krótkiego ramienia chromosomu 1. Region ten prawdopodobnie zawiera geny hamujące rozwój nowotworu (supresory).
- Delecja 11q: Utrata fragmentu długiego ramienia chromosomu 11. Jest to zmiana często występująca w guzach bez amplifikacji MYCN, ale również wiążąca się z agresywnym przebiegiem.
- Gain 17q: Zyskanie dodatkowego materiału genetycznego na chromosomie 17 jest najczęstszą anomalią genetyczną w neuroblastomie i silnym predyktorem niekorzystnego wyniku leczenia.
Najnowsze badania, prowadzone m.in. przez Memorial Sloan Kettering Cancer Center, Hospital Sant Joan de Déu, Barcelona, Ośrodki w Niemczech (Charité w Berlinie, Heidelberg), Dana-Farber/Boston Children’s Hospital rzucają również światło na rolę mechanizmów utrzymania telomerów. Komórki nowotworowe, aby stać się „nieśmiertelne”, muszą zapobiegać skracaniu się końcówek chromosomów (telomerów). W neuroblastomie dzieje się to poprzez nadekspresję telomerazy (często napędzaną przez MYCN) lub poprzez mutacje w genie ATRX. Odkryto również rzadkie, ale bardzo groźne mutacje w promotorze genu TERT, które wiążą się z wyjątkowo oporną na leczenie postacią choroby.
Obraz kliniczny i symptomatologia
Objawy neuroblastomy są „wielkim imitatorem” w pediatrii. Ponieważ guz wywodzi się z układu współczulnego, który unerwia całe ciało, lokalizacja ogniska pierwotnego może być bardzo różna, co determinuje objawy. Najczęstszą lokalizacją (ok. 65%) jest jama brzuszna, a konkretnie rdzeń nadnerczy, ale guzy mogą powstawać wzdłuż całego kręgosłupa: na szyi, w klatce piersiowej czy miednicy.1
Objawy wynikające z lokalizacji guza
- Jama brzuszna: Rodzice często zauważają powiększenie obwodu brzucha, wyczuwalny twardy guz, skarżą się na zaparcia u dziecka lub bóle brzucha. Guz może uciskać nerki lub naczynia nerkowe, co prowadzi do nadciśnienia tętniczego, rzadko wykrywanego rutynowo u małych dzieci.
- Klatka piersiowa: Guzy śródpiersia mogą osiągać duże rozmiary zanim dadzą objawy, takie jak przewlekły kaszel, duszność, świszczący oddech czy nawracające infekcje, wynikające z ucisku na drogi oddechowe.
- Odcinek szyjny: Guz zlokalizowany wysoko może uszkodzić splot nerwowy, prowadząc do tzw. zespołu Hornera. Jest to charakterystyczna triada objawów: opadnięcie powieki (ptosis), zwężenie źrenicy (miosis) i zapadnięcie gałki ocznej w głąb oczodołu (enophthalmos) po jednej stronie twarzy. Czasami towarzyszy temu brak potliwości na połowie twarzy (anhidrosis).
- Kanał kręgowy: Guzy rosnące przez otwory międzykręgowe do kanału kręgowego (tzw. guzy „klepsydrowe”) stanowią zagrożenie neurologiczne. Ucisk na rdzeń kręgowy może powodować bóle pleców, osłabienie siły mięśniowej nóg, utykanie, a w zaawansowanych przypadkach porażenia kończyn i zaburzenia oddawania moczu/stolca. Jest to stan nagły wymagający natychmiastowej interwencji.
Objawy przerzutowe
Niestety, u około 50% pacjentów w momencie diagnozy choroba jest już rozsiana.
- Układ kostny: Przerzuty do kości są bolesne. Dziecko może odmawiać chodzenia, budzić się w nocy z płaczem, utykać.
- Oczodoły: Przerzuty do kości czaszki i oczodołów dają bardzo specyficzny objaw tzw. „oczu szopa pracza” lub „krwiaków okularowych”. Są to zasinienia wokół oczu, często z towarzyszącym wytrzeszczem gałek ocznych (proptosis), co bywa mylnie brane za skutek urazu.
- Szpik kostny: Zajęcie szpiku prowadzi do wypierania prawidłowych krwinek, co objawia się bladością (anemia), skłonnością do siniaków i krwawień (małopłytkowość) oraz częstymi infekcjami (leukopenia).
- Skóra: W specyficznej dla niemowląt postaci 4S, na skórze mogą pojawiać się liczne, niebieskawe, twarde guzki, co obrazowo określa się mianem „zespołu jagodowego ciastka” (blueberry muffin syndrome).
Zespoły paraneoplastyczne
Neuroblastoma jest guzem czynnym hormonalnie, co może wywoływać objawy systemowe.
- Nadmierne wydzielanie katecholamin: Powoduje nadciśnienie, potliwość, kołatanie serca i drażliwość.
- Biegunki wodniste: Związane z wydzielaniem wazoaktywnego peptydu jelitowego (VIP) przez guz. Biegunki te są oporne na leczenie objawowe.
- Zespół opsoklonie-mioklonie (OMS): Fascynujące i rzadkie zaburzenie autoimmunologiczne, w którym układ odpornościowy, próbując walczyć z guzem, atakuje móżdżek dziecka. Objawia się to chaotycznymi, szybkimi ruchami gałek ocznych („tańczące oczy”), drżeniem mięśni i ataksją (niezbornością ruchową). Co paradoksalne, dzieci z OMS często mają neuroblastomę o korzystniejszej biologii i lepszym rokowaniu onkologicznym, jednak mogą borykać się z trwałymi następstwami neurologicznymi i rozwojowymi.
Ścieżka diagnostyczna i ocena zaawansowania
Prawidłowe postawienie diagnozy to proces wieloetapowy. W Polsce diagnostyka neuroblastomy odbywa się w wyspecjalizowanych ośrodkach onkologii i hematologii dziecięcej, zgodnie ze standardami Polskiego Towarzystwa Onkologii i Hematologii Dziecięcej (PTOHD) oraz wytycznymi europejskimi.
Badania laboratoryjne i biochemiczne
Podstawą jest badanie poziomu metabolitów katecholamin w dobowej zbiórce moczu. Komórki neuroblastomy produkują dopaminę, norepinefrynę i epinefrynę, które są metabolizowane do kwasu wanilinomigdałowego (VMA) i homowanilinowego (HVA). Ich podwyższony poziom obserwuje się u ponad 90% pacjentów i jest to czuły marker diagnostyczny. Dodatkowo we krwi oznacza się poziom enolazy swoistej dla neuronów (NSE), ferrytyny oraz dehydrogenazy mleczanowej (LDH). Wysoki poziom tych markerów (szczególnie ferrytyny i LDH) może sugerować dużą masę guza i gorsze rokowanie.
Obrazowanie molekularne i radiologiczne
Kluczowe dla oceny zasięgu choroby są badania obrazowe:
- Scyntygrafia MIBG (z użyciem 123I): Jest to „złoty standard” w diagnostyce neuroblastomy. Metajodobenzyloguanidyna (MIBG) to substancja chemicznie podobna do noradrenaliny, która jest aktywnie wychwytywana przez komórki guza. Podanie znacznika pozwala na zmapowanie całego ciała i wykrycie nawet drobnych przerzutów w kościach czy tkankach miękkich.
- PET-CT: U około 10% pacjentów guzy nie wychwytują MIBG. W takich przypadkach stosuje się pozytonową tomografię emisyjną (PET) z użyciem fluorodeoksyglukozy (FDG), która obrazuje metabolizm glukozy w komórkach nowotworowych.
- Tomografia Komputerowa (TK) i Rezonans Magnetyczny (MRI): Służą do precyzyjnej oceny guza pierwotnego, jego wielkości i stosunku do otaczających narządów oraz naczyń krwionośnych. Jest to kluczowe dla planowania operacji chirurgicznej. W systemie INRGSS ocenia się tzw. Image Defined Risk Factors (IDRF) – czynniki ryzyka widoczne w obrazie, np. czy guz „obrasta” aortę, co czyni go trudnym do usunięcia.
Ocena szpiku i histopatologia
Aby potwierdzić lub wykluczyć przerzuty do szpiku, wykonuje się biopsję aspiracyjną i trepanobiopsję, zazwyczaj z dwóch lub czterech miejsc (talerze kości biodrowej). Ostateczne rozpoznanie stawia się jednak na podstawie badania wycinka guza pobranego chirurgicznie. Patolog ocenia typ tkanki (klasyfikacja INPC) oraz wykonuje badania genetyczne (FISH, PCR) w celu oceny amplifikacji MYCN i innych aberracji.
Od diagnozy do planu działania
Zakończenie pełnego procesu diagnostycznego to dla rodziców jeden z najtrudniejszych, ale i najważniejszych momentów. Choć zebranie tak szczegółowych danych – od poziomu metabolitów w moczu , przez precyzyjne mapowanie ciała w scyntygrafii MIBG , aż po głęboką analizę genetyczną MYCN, ALK czy telomerów – wymaga czasu i wielu badań, jest to proces niezbędny do podjęcia właściwej walki.
Współczesna medycyna nie leczy już „neuroblastomy w ogóle”. Dzięki opisanym badaniom molekularnym i obrazowym lekarze zyskują pełny „dowód osobisty” nowotworu. To właśnie te unikalne cechy biologiczne guza pozwalają precyzyjnie określić, czy dziecko potrzebuje jedynie czujnej obserwacji ze względu na szansę na samoistną regresję, czy też wymaga najbardziej zaawansowanych protokołów leczenia agresywnych postaci choroby.
Zrozumienie biologii przeciwnika to pierwszy i kluczowy krok do zwycięstwa. Z kompletem tych informacji pacjent zostaje zakwalifikowany do konkretnej grupy ryzyka, co otwiera drogę do zaplanowania wieloetapowej terapii. W kolejnej części naszego kompendium przyjrzymy się szczegółowo strategiom leczenia – od precyzyjnej chirurgii i chemioterapii, po nowoczesną immunoterapię anty-GD2 i inhibitory celowane, które dają szansę na zdrowie dzieciom nawet z najbardziej skomplikowanymi postaciami neuroblastomy.
Co dalej? W kolejnej części naszego kompendium przyjrzymy się szczegółowo strategiom leczenia – od precyzyjnej chirurgii, przez chemioterapię, aż po przełomowe inhibitory celowane i immunoterapię anty-GD2